По Джину Д. Вилсону, Джеймсу Е. Гриффину Ш (JeanD. Wilson, JamesE. GriffinШ)
Внимание! В тексте устаревшая строго медицинская терминология!
Клинические проявления нетипичного хромосомного пола
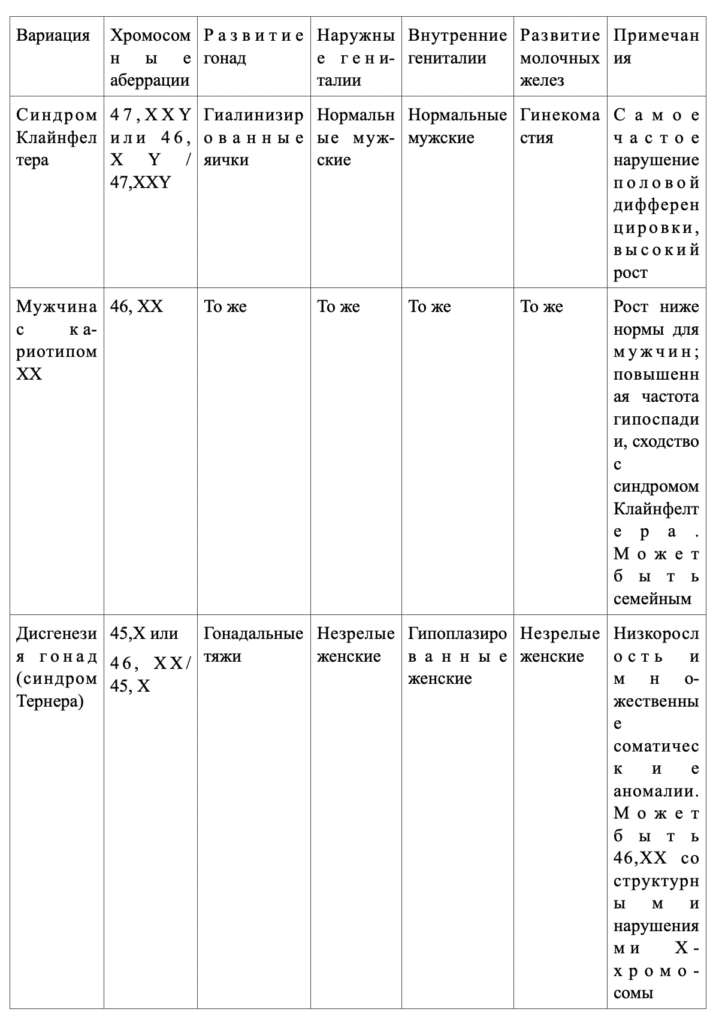
Синдром Клайнфелтера
Клинические проявления.Синдром Клайнфелтера характеризуется первичным гипогонадизмом (маленькие твердые яички), азооспермией, гинекомастией и повышенным уровнем гонадотропинов в плазме мужчин с двумя или более Х-хромосомами. Кариотип — чаще 47, XXY (классическая форма) или мозаицизм 46, XY/47, XXY. Этот синдром представляет собой самое распространенное нарушение половой дифференцировки и встречается с частотой примерно 1 случай на 500 мужчин.
В препубертатном возрасте у больных отмечают маленькие тестикулы, но в остальном они выглядят нормально. После полового созревания болезнь проявляется бесплодием, гинекомастией или иногда недостаточной андрогенизацией (табл.333-3). Постоянным признаком кариотипа 47, XXY является гиалинизация семенных канальцев и азооспермия. Яички небольшие, плотные, длиной менее 2см (всегда меньше 3,5см), что соответствует объему 2 мл (12 мл). Увеличение среднего роста определяется удлинением нижней части туловища. Гинекомастия появляется обычно в отрочестве и, как правило, с обеих сторон; молочные железы болезненны и могут увеличиваться до размеров, изменяющих фигуру (см. гл.332). От 30 до 50% больных страдают ожирением и варикозным расширением вен. Часто встречаются легкая психическая отсталость, трудности в социальной адаптации, нарушения функции щитовидной железы, сахарный диабет и заболевания легких. Риск возникновения рака молочных желез в 20 раз превышает таковой среди здоровых мужчин (но в 5 раз меньше такового у женщин). Для большинства больных характерна мужская психосексуальная ориентация, половая функция у них как у здоровых мужчин.
По результатам исследования хромосомного кариотипа в лейкоцитах периферической крови установлено, что мозаичным вариантом синдрома страдают около 10% больных. Частота этого варианта, по-видимому, занижена, так как хромосомный мозаицизм может иметь место только в тестикулах, а кариотип периферических лейкоцитов — оставаться нормальным. Мозаичная форма протекает обычно не столь тяжело, как вариант 47, XXY, и яички могут сохранять нормальные размеры (см. табл.333-3). Эндокринные нарушения также выражены слабее, а гинекомастия и азооспермия встречаются реже. Больше того, больные с мозаицизмом иногда могут сохранять фертильность. У некоторых из них из-за незначительности физических отклонений от нормы можно и не заподозрить правильного диагноза.
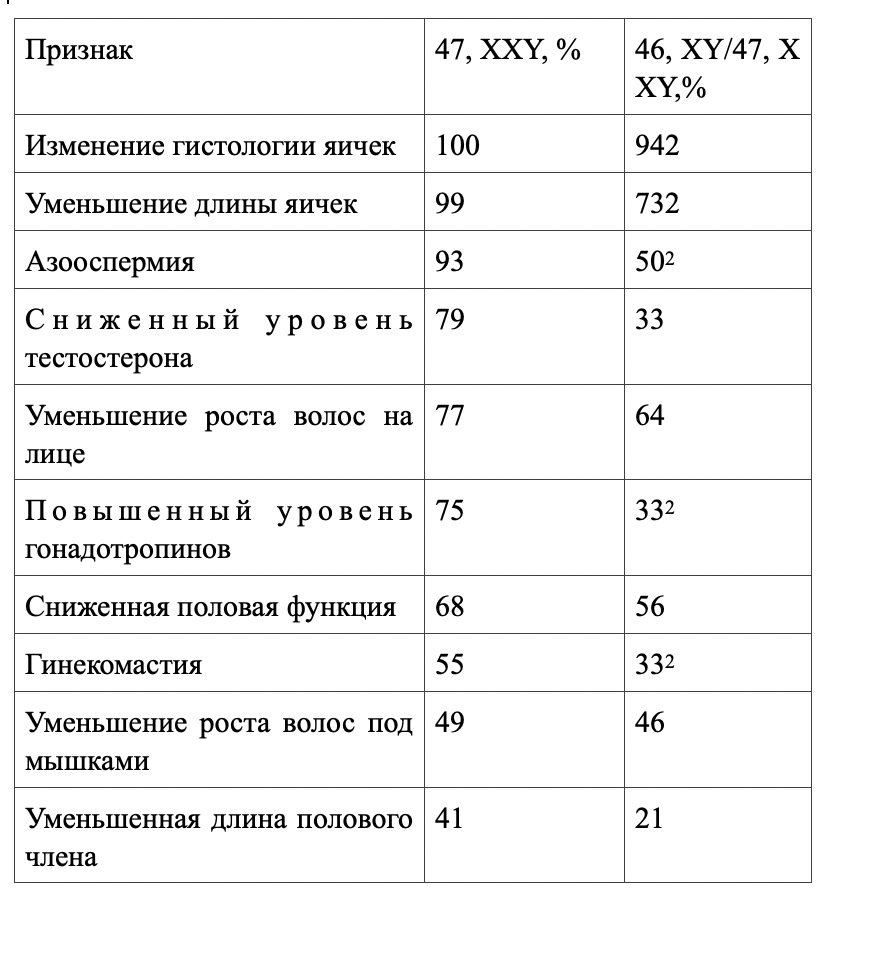
Характеристика больных с классическим и мозаичным вариантом синдрома Клайнфелтера (Таблица основана на результатах обследования 519 больных с кариотипом XX и 51 с кариотипом XY/XXY— больного. 2 Вероятность различий р <0,05 или еще выше. Из Gordon et al.)
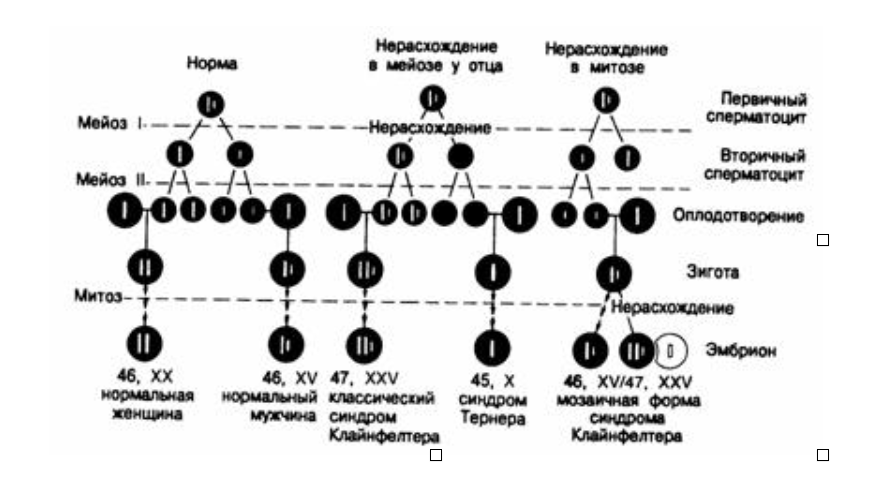
Схема нормального сперматогенеза и оплодотворения.
Показаны эффекты нерасхождения в мейозе и митозе, что приводит к формированию классического синдрома Клайнфелтера, синдрома Тернера и мозаичной формы синдрома Клайнфелтера. Схема не изменится, если нарушения будут происходить в процессе оогенеза.
Описано еще около 30 вариантов кариотипа при синдроме Клайнфелтера, как без мозаицизма (XXYY, XXXYи XXXXY), так и с мозаицизмом с сопутствующими структурными нарушениями Х-хромосомы или без них. Как правило, чем больше степень хромосомных нарушений (а при мозаичной форме — чем больше патологических клеточных линий), тем более тяжелы клинические проявления.
Патофизиология.Классическая форма обусловливается нерасхождением хромосом в мейозе в процессе гематогенеза (рис.333-2). Примерно в 40% случаев нерасхождение в мейозе происходит при сперматогенезе, а в 60%— при оогенезе. С увеличением возраста матери вероятность нерасхождения увеличивается. Мозаичную форму относят за счет нерасхождения хромосом в митозе после оплодотворения яйцеклетки; это нерасхождение может иметь место как в 46, XY-зиготе, так и в 47, XXY-зиготе. Двойной дефект (нерасхождение и в мейозе и в митозе) чаще всего служит причиной синдрома и объясняет тем самым, почему его мозаичная форма диагностируется реже, чем классическая.
Содержание фолликулостимулирующего (ФСГ) и лютеинизирующего гормона (ЛГ) в плазме обычно повышено; из-за постоянного дефекта семенных канальцев уровень ФСГ меньше перекрывается нормальными показателями и имеет большее диагностическое значение. Уровень тестостерона в плазме в среднем составляет половину нормального, но его колебания перекрываются нормальными. Среднее содержание эстрадиола в плазме по не совсем ясным причинам повышено. На ранних этапах заболевания яички могли бы секретировать большие количества эстрадиола вследствие повышенного уровня ЛГ в плазме, но в конце концов тестикулярная секреция эстрадиола (и тестостерона) снижается. Повышение содержания эстрадиола на поздних стадиях заболевания можно объяснить, вероятно, сочетанием уменьшения скорости его метаболического клиренса с ускорением конверсии тестостерона в эстрадиол вне железы. В результате как на ранних, так и на поздних стадиях проявляется та или иная степень недостаточной андрогенизации и избыточной феминизации. Феминизация, включая гинекомастию, зависит от относительного или абсолютного преобладания эстрогенов над андрогенами в крови, и у лиц с меньшим содержанием тестостерона и большим содержанием эстрадиола возрастает вероятность развития гинекомастии (см. гл.332). Повышение содержания гонадотропинов в плазме после введения рилизинг-гормона лютеинизирующего гормона (ЛГРГ) в постпубертатном возрасте более значительное, а нормальное ингибирующее действие тестостерона на гипофизарную секрецию ЛГ (отрицательная обратная связь) ослаблено. У больных с нелеченым синдромом Клайнфелтера может иметь место «реактивная патология гипофиза» в виде увеличения или деформации турецкого седла. Это объясняется, по-видимому, хроническим выпадением влияния гонад по механизму отрицательной обратной связи и гипертрофией гонадотрофов вследствие их стимуляции ЛГРГ. Возникает ли в таких случаях настоящая аденома, неизвестно.
Лечение.Восстановить фертильность при синдроме Клайнфелтера невозможно, а единственным эффективным способом коррекции гинекомастии является хирургическое удаление ткани молочных желез. Некоторым больным с недостаточной андрогенизцией помогает терапия андрогенами, но иногда она приводит к парадоксальному усилению гинекомастии, вероятно, за счет того, что увеличивает доступность субстратов для образования эстрогенов в периферических тканях. Андрогены следует применять в форме тестостерона ципионата или тестостерона энантата. При введении тестостерона уровень ЛГ в плазме, если и нормализуется, то лишь через несколько месяцев.
Синдром ХХ-мужчины
Кариотип 46, XX у фенотипических мужчин встречается с частотой приблизительно 1:20000 — 1:24000. У таких лиц все женские внутренние гениталии отсутствуют, и в психосексуальном плане они ощущают себя мужчинами. Действительно, признаки этого синдрома сходны с таковыми при синдроме Клайнфелтера: яички маленькие и плотные (обычно меньше 2см), часто наблюдается гинекомастия, азооспермия и гиалинизация семенных канальцев, половой член либо нормальных либо уменьшенных размеров. Концентрация тестостерона в плазме понижена, эстрадиола повышена, а содержание гонадотропинов достигает высокого уровня. Такие больные отличаются от больных с классическим синдромом Клайнфелтера только тем, что их рост в среднем ниже, чем у нормальных мужчин, психическое отставание встречается не чаще, чем в общей популяции, и увеличена частота гипоспадии.
Патогенез этого нарушения объясняют следующим образом:1) транслокацией части Y-хромосомы на Х-хромосому;2) мозаицизмом по Y-хромосоме в некоторых клеточных линиях или ранней потерей Y-хромосомы; 3) мутацией аутосомного гена и 4) делецией генетического вещества Х-хромосомы, в норме оказывающего отрицательный регуляторный эффект на развитие яичек. Однако ни одна из них не в состоянии полностью объяснить данное нарушение. Мозаицизм в большинстве случаев вряд ли имеет место, но все остальные процессы вполне возможны. Нельзя исключить и гетерогенную природу синдрома. Терапевтические мероприятия в таких случаях аналогичны таковым при синдроме Клайнфелтера.
Дисгенезия гонад (синдром Тернера)
Клинические проявления.Дисгенезия гонад характеризуется первичной аменореей, половым инфантилизмом, низкорослостью, множественными врожденными аномалиями и наличием гонадальных тяжей с обеих сторон у фенотипических женщин с каким-либо дефектом Х-хромосомы. Это состояние следует отличать:1) от смешанной дисгенезии гонад, при которой с одной стороны имеется яичко, а с другой — гонадальный тяж;2) от чистой дисгенезии гонад: в этом случае гонадальные тяжи с обеих сторон имеют место у лиц с нормальным кариотипом 46, XX или 46, XY, нормальным ростом и первичной аменореей;3) синдрома Нунан — аутосомно-доминантного нарушения у мужчин и женщин, характеризующегося складчатой кожей на шее, низкорослостью, врожденными пороками сердца, вальгусной деформацией предплечий и другими врожденными дефектами, несмотря на нормальные кариотип и гонады.
Частота дисгенезии гонад — 1:2500 новорожденных девочек. Диагноз ставят либо сразу после рождения по сопутствующим врожденным порокам, либо, что чаще, в пубертатном возрасте, когда врожденным аномалиям сопутствует аменорея. Дисгенезия гонад — самая распространенная причина первичной аменореи (около 30%). Больные не достигают полового созревания, наружные гениталии женского типа, но недоразвитые, так же как и молочные железы (в том случае, если больная не лечилась эстрогенами). Внутренние гениталии представлены инфантильными маточными трубами и маткой; в широких связках с обеих сторон присутствуют гонадальные тяжи. В процессе эмбриогенеза транзиторно появляются примордиальные зародышевые клетки, но они исчезают в результате ускоренной атрезии (см. гл.331). К возрасту ожидаемого полового созревания эти тяжи уже не содержат различимых фолликулов и яйцеклеток; в них присутствует фиброзная ткань, неотличимая от нормальной стромы яичников.
Сопутствующие соматические аномалии затрагивают в основном скелет и соединительную ткань. В младенческом возрасте болезнь диагностируют по наличию лимфатического отека кистей и стоп, складчатости шеи, низкой линии оволосения, избыточных кожных складок на затылке, щитообразной грудной клетке с широко расставленными сосками и малой массе тела при рождении. Кроме того, у больных характерное лицо с маленькой челюстью, эпикантус, низко расположенные или деформированные уши, рыбий рог и птоз. В 50% случаев отмечают укорочение IV пястных костей, а в 10—20%—коарктацию аорты. Рост у взрослых больных редко превышает 150см. Сопутствующие нарушения включают пороки развития почек, пигментированные родимые пятна, гипоплазию ногтей, склонность к кетозу, потерю слуха, необъяснимую гипертензию и аутоиммунные нарушения. Около 20% больных страдают гипотиреозом.
Патофизиология.Примерно у 50% больных обнаруживают кариотип 45. X, у 25% — мозаицизм без структурных нарушений (46, ХХ/45,X). а у остальных — структурные па-рушения Х-хромосомы с мозаицизмом или без него (см. гл.60). Вариант 45, Х обусловлен потерей хромосомы в процессе гаметогенсза у любого из родителей или с ошибкой митоза при одном из ранних делений оплодотворенной зиготы (см. рис.332-2). Низкорослость и другие соматические изменения являются следствием потери генетического материала с короткого плеча Х-хромосомы. Гонадальные тяжи образуются при потере генетического материала либо с длинного, либо с короткого плеча Х-хромосомы. У больных с мозаицизмом или структурными нарушениями Х-хромосомы изменения фенотипа занимают по тяжести промежуточное положение между теми, которые наблюдаются при варианте 45,Х, и нормой. У некоторых больных с гипертрофией клитора, помимо Х-хромосомы, присутствует фрагмент еще какой-то хромосомы, предположительно — аномальной Y-хромосомы. В редких случаях сбалансированная Х-аутосомная транслокация может обусловить семейную передачу дисгенезии гонад (см. гл.60).
Раньше для выявления нарушений Х-хромосом исследовали половой хроматин. Половой хроматин (тельца Барра) у здоровых женщин — продукт инактивации одной из двух Х-хромосом; женщин с хромосомным набором,45,X, подобно нормальным мужчинам, относили к группе хроматинотрицательных. Однако хроматннотрицательными являются лишь около 50% больных с дисгенезией гонад (больные с кариотипом 45,Х и с наиболее выраженным мозаицизмом и структурными нарушениями). Поэтому для установления диагноза и идентификации больных с элементами Y-хромосомы и высоким риском возникновения злокачественных опухолей в гонадальных тяжах необходим анализ кариотипа.
В период ожидаемого полового созревания оволосение подмышек и лобка скудное, молочные железы неразвиты, менструаций нет. Содержание ФСГ в сыворотке, повышенное в младенчестве, в детстве снижается до нормы, а в возрасте 9—10 лет возрастает до уровня, характерного для кастратов. В это время содержание ЛГ в сыворотке также повышено, а уровень эстрадиола в плазме снижен (менее 10 пг/мл). Примерно у 2%>больных с вариантом 45.Х и у 12% больных с мозаициэмом в яичниках сохраняется достаточное число фолликулов, чтобы иногда возникали менструации. Больше того, у лип с минимальными повреждениями иногда возможна беременность. Однако продолжительность детородного периода у таких больных невелика.
Лечение.В ожидаемое время полового созревания следует начать заместительную терапию эстрогенами, чтобы индуцировать развитие молочных желез, половых губ, влагалища, матки и маточных труб (см. гл.331). В первый год лечения эстрадиолом скорость роста тела в длину и созревания костей примерно удваивается, но окончательный рост больных редко достигает ожидаемого (см. гл.331). Лечение гормоном роста не приносит успеха. У больных с вариантом 45, Х опухоли гонад встречаются редко, но у некоторых лиц с мозаицизмом по Y-хромосоме они возникают. Поэтому гонадальные тяжи следует удалять в любом случае при наличии признаков вирилизации пли при обнаружении линии клеток, содержащих Y-хромосому.
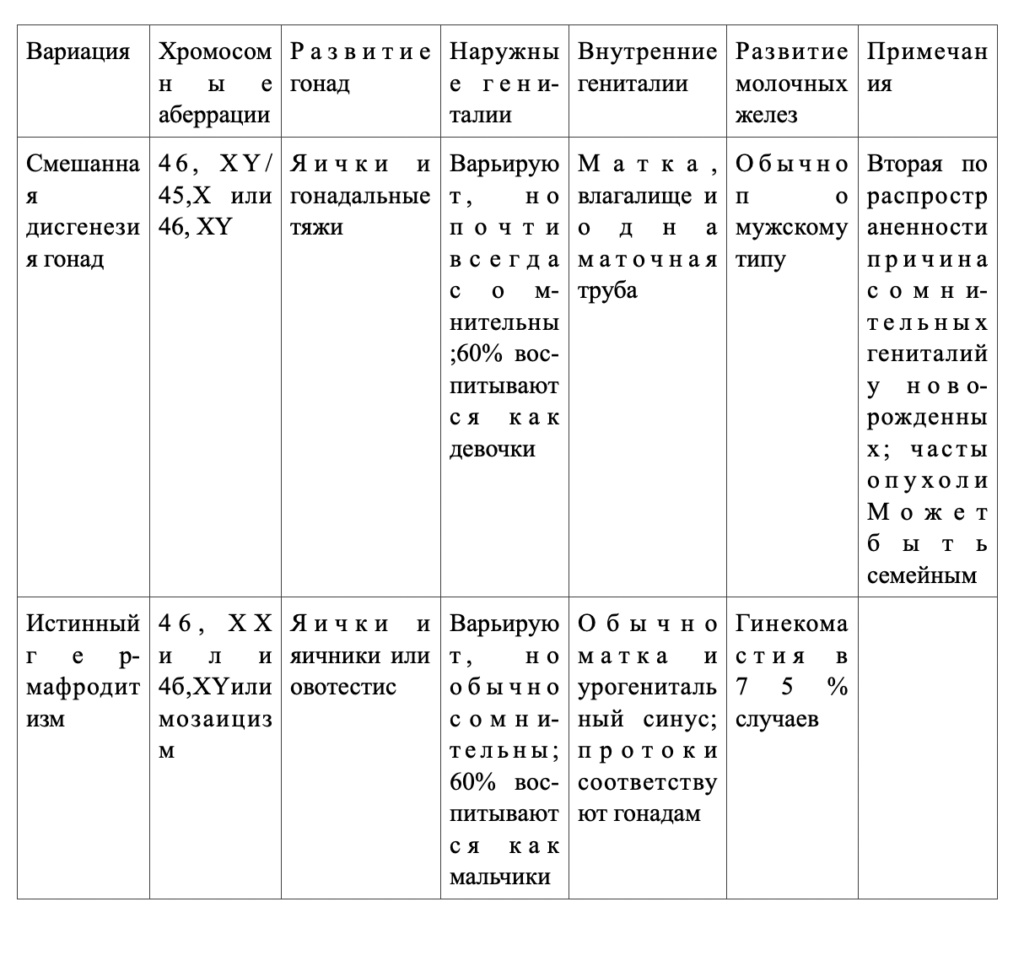
Смешанная дисгенезия гонад
Клинические проявления.Смешанная дисгенезия гонад —это состояние, при котором у фенотипических мужчин или женщин с одной стороны имеется яичко, а с другой — гонадальный тяж. У большинства больных обнаруживается мозаицизм 45, Х/46,XY, по клинические проявления выходят за рамки определяемых этой аберрацией хромосом. Частота синдрома неизвестна, но, по данным большинства клиник, это вторая по частоте (после врожденной гиперплазии надпочечников) причина амбисексуальности гениталий у новорожденных.
Примерно 60% больных считают девочками, а большинство фенотипических мальчиков при рождении не полностью вирилизированы. У большинства обнаруживают амбисексуальные гениталии, включая несколько увеличенный половой член, урогенитальный синус и в различной степени сращенные в мошонку половые губы. Яичко у большинства больных расположено интраабдоминально; лиц с яичком в паху или в мошонке считают мальчиками. Почти всегда есть матка, влагалище и по крайней мере одна маточная труба.
До пубертатного возраста яичко кажется относительно нормальным. В постпубертатном возрасте оно содержит множество зрелых клеток Лейдига, но семенные канальцы лишены зародышевых элементов и содержат лишь клетки Сертолн. Гонадальный тяж — тонкое бледное удлиненное образование, расположенное либо в широкой связке, либо на тазовой стенке, состоит из стромы яичника. У больных пубертатного возраста яичко секретирует андрогены и происходит как вирилизация, так и увеличение размеров полового члена. Феминизация встречается редко, но при ее наличии следует заподозрить секрецию эстрогенов опухолью гонад.
Примерно у 30% больных отмечают соматические проявления 45, Х-дисгенезии гонад — низкую заднюю линию оволосения, щитообразную грудную клетку, множественные пигментированные родимые пятна, вальгусную де4юрмацию предплечий, складчатость шеи и низкорослость (рост менее 150см).
Практически все больные хроматинотрицательны. При обследовании группы больных у 60% был обнаружен кариотип 45, X/46.XY, у остальных — кариотип 46, XY, но частота мозаицизма могла быть заниженной или ограниченной лишь некоторыми линиями клеток. Причину мозаицизма 45, Х/46,XY лучше всего объясняет утрата Y-хромосомы на ранних стадиях митотического деления XY-зиготы, подобная постулируемой потере Х-хромосомы при мозаицизме 46, XY/47, XXY, показанной на рис.333-2.
Патофизиология.Предполагают, что 46, XY-клеточная линия стимулирует дифференцировку яичка, тогда как 45, Х-линия обусловливает развитие контралатерального гонадального тяжа, но реальное сопоставление кариотипическнх и фенотипических проявлений не подтверждает такой зависимости. Больше того, отсутствует корреляция между процентом культивируемых клеток крови или кожи, содержащих 45, Х или 46. XY, и степенью развития гонад или соматическими аномалиями.
Как маскулинизация, так и регрессия мюллеровых протоков inuteroосуществляется не полностью. Поскольку в пубертате клетки Лейдига функционируют нормально, недостаточная внутриутробная вирилизация может быть следствием запаздывания развития яичка, клетки Лейдига в котором в конце концов приобретают способность к нормальному функционированию. Возможно также, что яичко плода просто не способно синтезировать нужное количество вещества, ингибирующего мюллеровы протоки, и андрогенов.
Лечение.Следует отметить, что у детей старшего возраста и взрослых лиц, у которых пол зафиксирован до установления диагноза, возможно появление в гонадах опухоли. Общая частота таких опухолей составляет 25%. Семиномы встречаются чаще, чем гонадобластомы, причем опухоли могут возникать и до полового созревания. Этому чаще подвержены больные с женским фенотипом, лишенные соматических признаков типичной 45,Х-дисгенезии гонад, интраабдоминальные яички поражаются чаще, чем гонадальные тяжи. Когда у больных с женским фенотипом диагноз установлен, следует провести эксплоративную лапаротомию и профилактическую гонадэктомию, как потому, что опухоли гонад могут развиваться в детстве, так и потому, что яичко в пубертате секретирует андрогены и вызывает тем самым вирилизацию. С целью индуцировать и поддерживать феминизацию таким больным, как и лицам с дисгенезией гонад, затем назначают эстрогены.
Лечение больных с мужским фенотипом, у которых диагноз установлен в старшем детском возрасте или в зрелые годы, затруднено. Фенотипические мужчины со смешанной дисгенезией гонад бесплодны (в яичках отсутствуют зародышевые элементы), и у них также повышен риск появления опухолей гонад. В каких же случаях можно без опасений сохранять яичко? Как правило, нужно учитывать следующее: опухоли развиваются в мошоночных гонадальных тяжах, но не в расположенных в мошонке яичках; опухоли, появляющиеся в неопущенных яичках, всегда связаны со структурами ипсилатерального мюллерова протока; опухоли в гонадальных тяжах всегда связаны с опухолями в контралатеральных интраабдоминальных яичках. Поэтому рекомендуется удалять все гонадальные тяжи, сохранять яички, находящиеся в мошонке, и удалять интраабдоминальные яички, кроме тех случаев, когда их удается низвести в мошонку и когда они не связаны со структурами ипсилатерального мюллерова протока. При проведении реконструктивных операций на половом члене необходимо учитывать характер дефекта.
Если диагноз установлен в раннем детстве и гениталии амбисексуальны, чаще выбирают женский пол. Позднее можно произвести резекцию увеличенного полового члена и гонадэктомию (обычно сразу). Если же выбирают мужской пол, то при решении вопроса об удалении яичка я детстве пользуются теми же критериями, что и у взрослых мужчин.
Истинный гермафродитизм
Клинические проявления.Истинный гермафродитизм — это состояние, при котором у больного есть и яичники и яички или гонады с гистологическими особенностями того и другого пола (овотестис). Подтвердить диагноз можно лишь в том случае, если при гистологическом исследовании обнаружен гонадный эпителий обоих типов (обнаружить только строму яичника без ооцитов недостаточно). Частота встречаемости синдрома неизвестна, но в литературе описано более 400 случаев. Больных подразделяют на три группы:
1)у 20% с обеих сторон присутствует и тестикулярная, и яичниковая ткань (овотестис);
2)у 40% на одной стороне имеется овотестис, а на другой — либо яичник, либо яичко;
3)у остальных на одной стороне присутствует яичко, а на другой — яичник.
Наружные половые органы у больных находятся на различных стадиях перехода от мужских к женским. Две трети достаточно маскулинизированных больных (их около 60%) воспитывают как мальчиков. Однако нормальные мужские наружные половые органы имеют меньше 10% больных; у большинства отмечают гипоспадию и более чем у 50%— неполное сращение половых губ в мошонку. У 60% лиц с женским фенотипом увеличен клитор, и у большинства имеется урогенитальный синус. Дифференцировка внутренних протоков обычно соответствует прилежащей гонаде. Хотя у яичка обычно развит придаток, семявыносящий проток полностью формируется лишь у 30% больных. Среди лиц с овотестис у 75% имеется придаток яичка, у 60% — маточная труба. Матка обычно гипоплазирована или имеет один рог. Яичники, как правило, занимают нормальное положение, но яички или овотестис могут обнаруживаться на любом уровне вдоль пути опущения яичка в эмбриогенезе и часто сочетаются с паховой грыжей. У 30% больных тестикулярная ткань локализуется в мошонке или губномошоночной складке, у 30% — в паховом канале, у остальных — в полости живота.
Период полового созревания характеризуется феминизацией и вирилизацией той или иной степени; у 75%) больных появляется гинекомастия и примерно у 50% — менструации. У лиц с мужским фенотипом менструации проявляются в виде циклической гематурии. Овуляция происходит примерно у 25% больных — чаще, чем сперматогенез. У лиц с мужским фенотипом овуляция может проявляться болями в яичках. Описаны фертильные индивиды с женским фенотипом, у которых были удалены овотестис, а также «мужчина», имевший двоих детей. Врожденные дефекты других систем встречаются редко.
Патофизиология.Примерно у 60% больных кариотип 46, XX, у 10% — 46, XY, а у остальных — хромосомный мозаицизм, при котором присутствует клеточая линия с Y-хромосомой. Механизм, определяющий такое развитие гонад, неизвестен. Полагают (хотя это и не доказано имеющимися методами кариотипирования), что в данном случае присутствует достаточное количество генетического материала Y-хромосомы (вследствие транслокации, нерасхождения или мутации), чтобы индуцировать развитие тестикулярной ткани. В редких случаях поражаются многие сибсы с кариотипом 46, XX, что обусловлено, вероятно, присутствием аутосомно-рецессивного гена или общей транслокацией.
Поскольку в яичниках более 25% больных содержатся желтые тела, можно заключить, что у таких индивидов нормально функционирует женская нейроэндокринная система. Феминизация (гинекомастия и менструации) обусловлена секрецией эстрадиола имеющейся яичниковой тканью. У маскулинизированных индивидов секреция андрогенов преобладает над секрецией эстрогенов, и у некоторых из них вырабатываются сперматозоиды.
Лечение.Если диагноз установлен у новорожденного или ребенка младшего возраста, выбор пола зависит от анатомических признаков. У детей старшего возраста и у взрослых следует удалять гонады и их внутренние протоки, противоречащие преобладающему фенотипу (и полу воспитания), и в необходимых случаях соответственно изменять наружные половые органы. Хотя при истинном гермафродитизме опухоли гонад встречаются редко, у лиц с XY-клеточной линией диагностировали гонадобластомы. Поэтому при решении вопроса о сохранении ткани гонад надо учитывать возможность появления в них опухоли.
Нарушения гонадного пола
О нарушении гонадного пола говорят в том случае, когда дифференцировка гонад не соответствует хромосомному полу, т. е. хромосомный пол не соответствует гонадному ифенотипическому полу.
Чистая дисгенезия гонад
Клинические проявления.Чистая дисгенезия гонад — это нарушение, при котором индивиды с женским фенотипом, половые органы которых, в том числе гонады, идентичны таковым у лиц с дисгенезией гонад (двусторонние гонадальные тяжи, инфантильные матка и маточные трубы и половой ин4>антилизм), имеют нормальный рост, нормальный кариотип (46, XX или 46, XY), при этом у них практически отсутствуют врожденные аномалии. Это состояние встречается в 10 раз реже, чем дисгенезия гонад. С генетической точки зрения, оно отлично от дисгенезии гонад, но дифференцировать чистую дисгенезию гонад от дисгенезии гонад с минимальными соматическими аномалиями по клиническим признакам невозможно. Больные, как правило, высокого роста (иногда более 170см). Дефицит эстрогенов варьирует от резко выраженного, характерного для типичной 45, Х-дисгенезии гонад, до незначительного. В последнем случае у больных до некоторой степени развиты молочные железы, бывают менструации, хотя менопауза наступает довольно рано. Примерно у 40% больных отмечают определенной степени феминизацию. Подмышечное и лобковое оволосение скудное, а внутренние половые органы представлены лишь производными мюллеровых протоков.
В гонадальных тяжах могут развиваться опухоли, особенно дисгерминомы или гоиадобластомы (при кариотипе 46, XY). Такие опухоли часто сопровождаются признаками вирилизации или появлением плюс-ткани в области таза.
Патофизиология.Хотя термин «чистая дисгенезия гонад» использовали и для описания случаев хромосомного мозаицизма, мы относим его лишь к немозаичным случаям кариотипа 46, XX или 46, XY. (Мозаицизм представляет собой варианты дисгенезии гонад или смешанной дисгенезии гонад, обсуждаемых выше.) Основанием для такого разграничения служит тот факт: что как XX-, так и XY-варианты данного синдрома могут быть следствием мутаций одиночного гена. Описаны семьи, в которых кариотип 46, XX выявляли у нескольких снбсов; это часто наблюдалось при браках между кровными родственниками, что указывает на аутосомно-рецессивный характер наследования. Отмечали и семейные случаи варианта 46, XY; иногда мутация передается, по-видимому, как сцепленный с Х-хромосомой рецессивный признак, тогда как в других семьях распространенность этого синдрома соответствует аутосомно-рецессивному наследованию признака, проявляющегося только у мужчин. При обоих формах (46, XX и 46, XY) мутация препятствует дифференцировке соответственно яичников или яичек; механизм этого неясен. Если гонады не развиваются, формируется женский фенотип. Как и у всех лиц с нефункционирующими гонадами, секреция гонадотропинов повышена, а эстрогенов снижена.
Лечение.Лечение больных с дефицитом эстрогенов аналогично таковому при дисгенезии гонад. Заместительную терапию эстрогенами начинают в срок ожидаемого полового созревания и продолжают на протяжении всего периода зрелости (см. гл.331). Больным с кариотипом 46, XY после установления диагноза следует провести эксплоративную операцию и удалить гонадальные тяжи в связи с высокой частотой появления у них опухолей гонад. Показанием к немедленной операции служит проявление признаков вирилизации. Естественное развитие опухолей гонад при данном синдроме остается неясным, но прогноз после их хирургического удаления обычно благоприятный.
Синдром отсутствия тестикул (анорхия, тестикулярная регрессия, агенезия гонад, агонадизм)
Клинические проявления.Индивиды с кариотипом 46, XY, у которых отсутствуют или имеются лишь рудиментарные яички, но на каком-то этапе внутриутробной жизни появляются несомненные признаки эндокринной функции этих желез (например, обязательная регрессия мюллеровых протоков и секреция тестостерона), могут иметь различный фенотип. Это достаточно редко встречающееся нарушение следует отличать от чистой дисгенезии гонад, при которой нет признаков, указывающих на функционирование гонад в процессе эмбрионального развития. Клинически синдром проявляется по-разному — полным отсутствием вирилизации, неполной вирилизацией наружных половых органов различной степени или нормальным мужским фенотипом, за исключением двусторонней анорхии.
Самая чистая форма патологии — это лица с женским фенотипом и кариотипом 46, XY. У них нет тестикул, выражен половой инфантилизм и отсутствуют как производные мюллеровых протоков, так и акцессорные органы мужской репродуктивной системы. Такие больные отличаются от лиц с 46, XY-формой чистой дисгенезии гонад тем, что у них не удается обнаружить никаких остатков гонад: ни гонадальных тяжей, ни производных мюллеровых протоков. Тестикулярная недостаточность должна возникать на этапе между началом образования вещества, ингибирующего мюллеровы протоки, и секрецией тестостерона, т. е. уже после развития семенных канальцев, но перед началом функционирования клеток Лейдига.
У других больных клиническая картина свидетельствует о более позднем проявлении тестикулярной недостаточности в процессе внутриутробного развития, и у них могут возникать трудности при выборе пола. В некоторых случаях недостаточность регрессии мюллеровых протоков может быть выражена сильнее, чем недостаточность секреции тестостерона, но полного развития мюллеровых структур не происходит никогда. У лиц с более значительной вирилизацией наружные половые органы имеют мужской фенотип, но могут одновременно присутствовать и рудиментарные яйцеводы и семявыносящие протоки.
Выделяют также синдром двусторонней анорхии у лиц с мужским фенотипом. При этом у больных отсутствуют мюллеровы структуры и гонады, но система вольфовых протоков и наружные половые органы развиты по мужскому тину. Наличие микропениса означает, что недостаточность опосредованного андрогенами роста полового члена возникает на поздних стадиях эмбриогенеза, уже после завершения анатомического формирования мужской уретры. После ожидаемого времени полового созревания у одних больных развивается постоянная гинекомастия, у других — нет.
Патофизиология.Патогенез болезни неясен. Регрессия яичек могла бы определяться мутантным геном, тератогеном или травмой. Описаны множественные случаи агонадиз-ма в одной и той же семье, причем у одних больных нарушение было односторонним, а у других —двусторонним.
Количественная динамика секреции половых стероидов изучена недостаточно. У двух больных с женским фенотипом и первичной аменореей, половым инфантилизмом и отсутствием внутренних половых органов кинетика андрогенов и эстрогенов была сходной с таковой при дисгенезии гонад; скорость продукции эстрогенов была низкой, секрецию тестостерона вообще не удалось обнаружить, что подтверждает функциональное, а не только анатомическое отсутствие яичек. У одного больного с мужским фенотипом и двусторонней анорхией продукция тестостерона и эстрогенов осуществлялась лишь за счет их периферического образования из андростендиона плазмы. Однако у некоторых больных, у которых при лапаротомии не удавалось обнаружить яички, уровень тестостерона в крови превышал таковой у кастрированных индивидов; вероятно, гормон продуцировался остатками яичек.
Лечение.Лиц с половым инфантилизмом и женским фенотипом следует лечить так же, как больных с дисгенезией гонад, т. с. им необходимо вводить эстрогены в количестве, способном вызвать развитие молочных желез и характерные для женщины соматических изменения. В случае любых проявлений сопутствующей агенезии влагалища показаны хирургические или консервативные методы. Подобно этому, лицам с мужским фенотипом и анорхией следует вводить андрогены в количестве, которое могло бы обеспечить развитие нормальных мужских вторичных половых признаков. Больным с неполной вирилизацией или амбисексуальным развитием наружных половых органов требуется индивидуальный подход к решению вопроса о необходимости хирургического лечения, помимо гормонального лечения в срок ожидаемого полового созревания.



